Neuron Available online 26 April 2023
Microglial-to-neuronal CCR5 signaling regulates autophagy in neurodegeneration (神経変性におけるオートファジーを制御するミクログリア-ニューロンCCR5シグナル)
Beatrice Paola Festa 125, Farah H. Siddiqi 125, Maria Jimenez-Sanchez 135, Hyeran Won 14, Matea Rob 12, Alvin Djajadikerta 12, Eleanna Stamatakou 12, David C. Rubinsztein 126
1Department of Medical Genetics, Cambridge Institute for Medical Research (CIMR), CB2 0XY Cambridge, UK2UK Dementia Research Institute, Cambridge Institute for Medical Research (CIMR), CB2 0XY Cambridge, UK
Received 24 August 2022, Revised 13 February 2023, Accepted 7 April 2023, Available online 26 April 2023.
Abstract
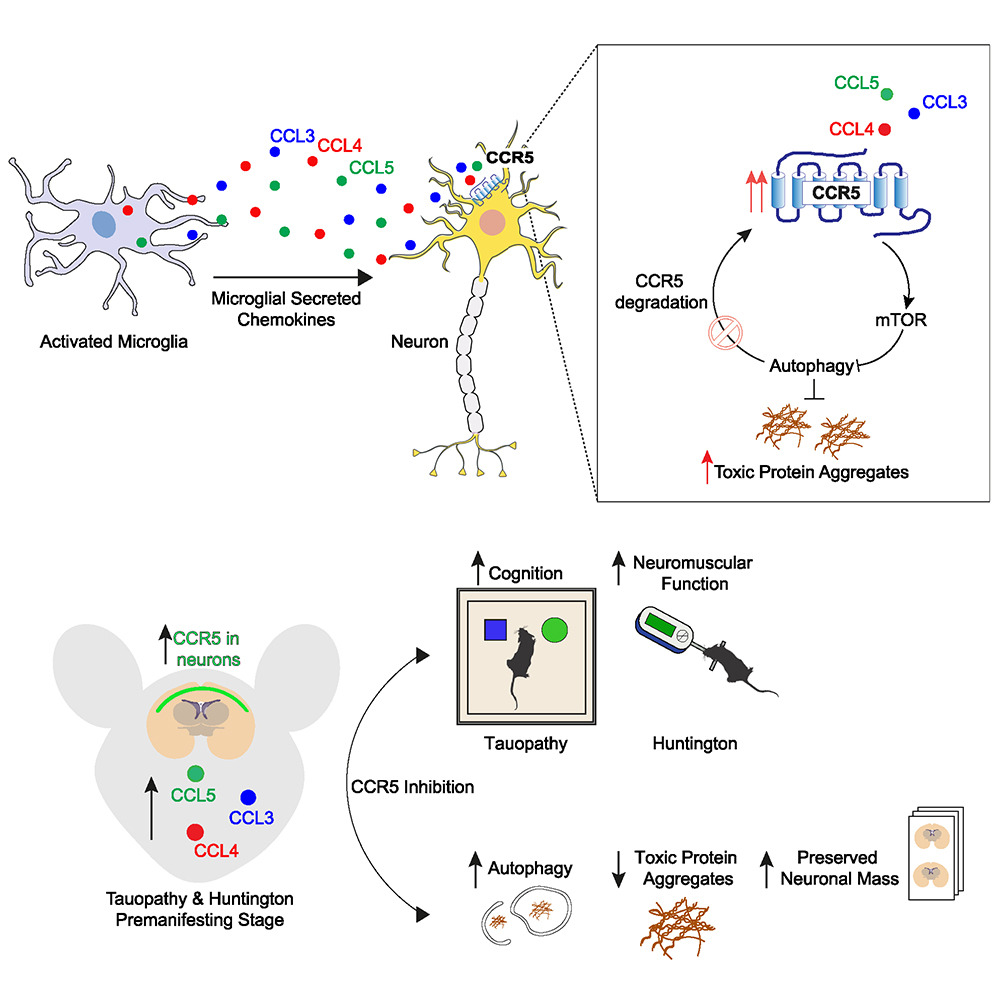
In neurodegenerative diseases, microglia switch to an activated state, which results in excessive secretion of pro-inflammatory factors. Our work aims to investigate how this paracrine signaling affects neuronal function. Here, we show that activated microglia mediate non-cell-autonomous inhibition of neuronal autophagy, a degradative pathway critical for the removal of toxic, aggregate-prone proteins accumulating in neurodegenerative diseases. We found that the microglial-derived CCL-3/-4/-5 bind and activate neuronal CCR5, which in turn promotes mTORC1 activation and disrupts autophagy and aggregate-prone protein clearance. CCR5 and its cognate chemokines are upregulated in the brains of pre-manifesting mouse models for Huntington’s disease (HD) and tauopathy, suggesting a pathological role of this microglia-neuronal axis in the early phases of these diseases. CCR5 upregulation is self-sustaining, as CCL5-CCR5 autophagy inhibition impairs CCR5 degradation itself. Finally, pharmacological or genetic inhibition of CCR5 rescues mTORC1 hyperactivation and autophagy dysfunction, which ameliorates HD and tau pathologies in mouse models.
神経変性疾患では、ミクログリアが活性化状態に切り替わり、その結果、炎症性因子が過剰に分泌される。我々の研究は、このパラクリンシグナル伝達が神経機能にどのような影響を及ぼすかを調べることを目的としている。ここでは、活性化したミクログリアが、神経変性疾患において蓄積される有毒で凝集しやすいタンパク質の除去に重要な分解経路である、神経細胞のオートファジーを細胞非自律的に阻害することを示す。われわれは、ミクログリア由来のCCL-3/-4/-5が神経細胞CCR5と結合して活性化し、その結果、mTORC1の活性化が促進され、オートファジーと凝集しやすいタンパク質の除去が阻害されることを発見した。CCR5とその同族ケモカインは、ハンチントン病(HD)とタウオパチーの発症前モデルマウスの脳で発現が上昇しており、これらの疾患の初期段階において、このミクログリア-ニューロン軸が病理学的役割を果たしていることを示唆している。CCL5-CCR5オートファジー阻害はCCR5の分解そのものを障害するため、CCR5の上昇は自己持続的である。最後に、CCR5の薬理学的あるいは遺伝学的阻害は、mTORC1の亢進とオートファジーの機能不全を解除し、マウスモデルにおけるHDとタウの病態を改善する。
Keywords: CCL5; CCR5; Huntington’s disease; Tau; autophagy; dementia; mTORC1; maraviroc; microglia; neuroinflammation.
Highlights
- Activated microglia mediate non-cell-autonomous inhibition of neuronal autophagy
- Microglial-derived CCL-5/-4/-3 activate neuronal CCR5 to inhibit neuronal autophagy
- CCR5 and CCL-3/-4/-5 are increased in Huntington’s disease and tauopathy mouse brains
- Pharmacological or genetic inhibition of CCR5 rescues neurodegeneration in mice
ハイライト
・活性化したミクログリアは、神経細胞のオートファジーを細胞非自律的に阻害する。
・ミクログリア由来のCCL-5/-4/-3は神経細胞のCCR5を活性化し、神経細胞のオートファジーを阻害する
・CCR5とCCL-3/-4/-5はハンチントン病とタウオパチーマウスの脳で増加している。
・CCR5を薬理学的あるいは遺伝学的に阻害すると、マウスの神経変性が回復する。
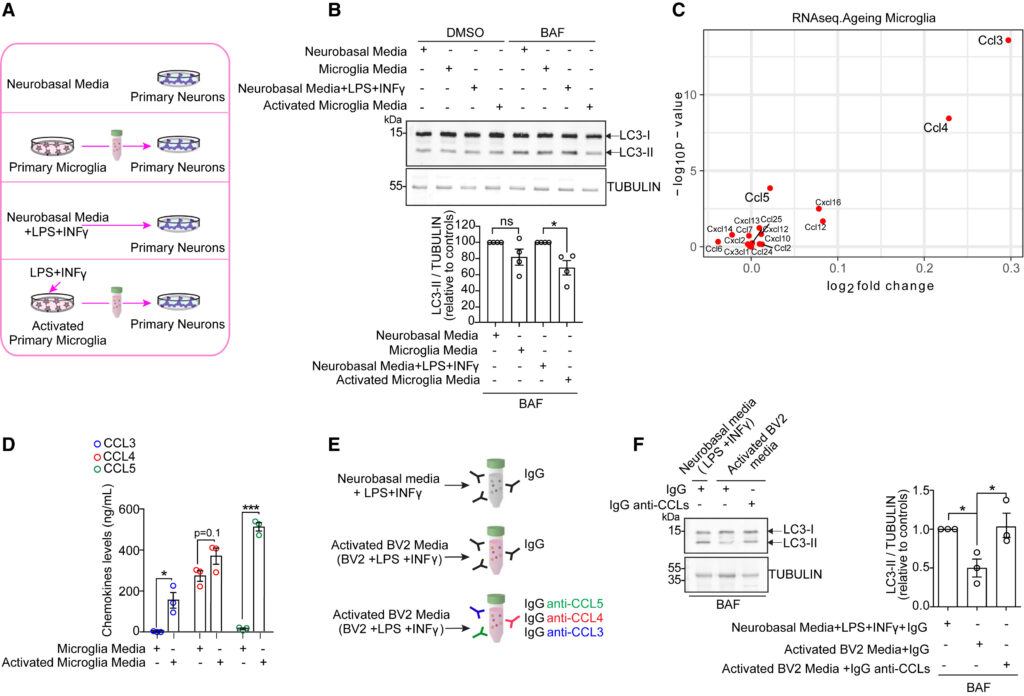
(A) Experimental conditions analyzed in (B). Mouse primary neurons were treated with conditioned media from primary microglia cultured in basal conditions or previously activated by LPS (10 ng/mL) + INFγ (100 ng/mL) for 24 h. Neurons alone cultured in neurobasal media with or without LPS+INFγ were used as controls.
(B) Western blotting and densitometry analyses of LC3-II protein levels in mouse primary neurons treated as described in (A) and incubated in DMSO or 400 nM bafilomycin A1 (BAF) for 8 h before being lysed. TUBULIN was loading control. Plots represent mean ± SEM (n = 4 independent experiments). Two-tailed, one-sample t test ∗p < 0.05, ns, not significant.
(C) Differential expression of chemokines in microglia from young and old mice. Scatterplot of −log10 p value against log2 fold-change from young to old. Data from Ximerakis et al.12
(D) ELISA measurement of CCL3, CCL4, and CCL5 levels in conditioned media from mouse primary microglia cultured in basal conditions or activated by LPS+INFγ. Plots represent mean ± SEM (n = 3 independent experiments). Two-tailed, unpaired t test ∗p < 0.05, ∗∗∗p < 0.001.
(E) Schematic of conditioned media used in experiments in (F). CCL3, CCL4, and CCL5 were immunodepleted from conditioned media derived from BV2 cells, previously activated by LPS (10 ng/mL) +INFγ (100 ng/mL), using specific IgG-anti-CCL3/-4/-5. Neurobasal media supplemented with LPS+INFγ or conditioned media from activated BV2 cells were incubated with non-specific IgG and used as controls.
(F) Western blotting and densitometry analyses of LC3-II levels in mouse primary neurons treated with CCL-3/-4/-5-immunodepleted conditioned media from activated BV2 cells or activated BV2 and neurobasal media treated as described in (E) and incubated in 400 nM BAF for 8 h before being lysed. TUBULIN was loading control. Plots represent mean ± SEM (n = 3 independent experiments). Two-tailed, paired t test ∗p < 0.05. See also Figure S1.
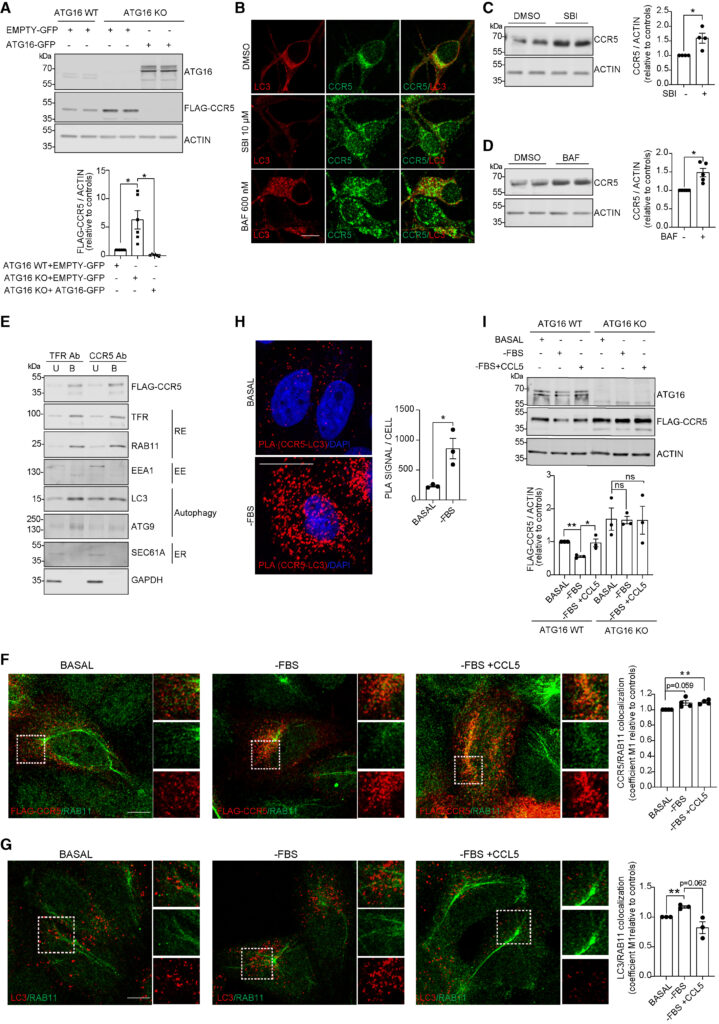
HeLa cells were transfected with FLAG-CCR5 constructs for 48 h in all experiments described below.
(A) Western blotting of FLAG-CCR5 and ATG16 and densitometry analyses of FLAG-CCR5 levels in ATG16 WT and KO HeLa cells transfected with Empty-GFP or ATG16-GFP construct for 48 h. GAPDH was loading control. Plots represent mean ± SEM (n = 6 independent experiments). Two-tailed, paired Student’s t test ∗p < 0.05.
(B–D) Mouse primary neurons were cultured in presence of DMSO or 10 μM SBI-0206965 (SBI) or 600 nM BAF for 8 h before being fixed for immunostaining or lysed. (B) Representative confocal micrographs of CCR5 (green) and LC3 (red) structures. Scale bars, 10 μm. (C and D) Western blotting and densitometry of CCR5 levels. ACTIN was loading control. Plots represent mean ± SEM (n = 4–5 independent experiments). Two-tailed, one-sample t test ∗p < 0.05.
(E) HeLa cells were starved for 1 h, loaded with ferrofluid conjugated with anti-TfR Ab or anti-CCR5 Ab for 1 h, and chased for 15 min in starvation medium. The cells were then fragmented and membranes containing ferrofluid conjugated with anti-TfR or anti-CCR5 Ab (B = bound) or not containing ferrofluid (U = unbound) were separated and processed for immunoblotting.
(F and G) HeLa cells were cultured in basal conditions or with FBS-depleted media in presence or absence of CCL5 (50 nM) for 4 h before fixation. Representative confocal micrographs and quantification of cells immunostained with anti-FLAG (red) and anti-RAB11 (green) in (F) or anti-RAB11 (green) and anti-LC3 (red) antibody in (G). High magnification insets are shown. Confocal micrographs of separate channels are included in Figures S7I and S7J. Plots represent mean ± SEM. (n = 3 independent experiments, n = 5–7 randomly selected fields/condition, each containing ∼ 10 cells) Two-tailed, paired Student’s t test ∗p < 0.05, ∗∗p < 0.01, ns not significant. Scale bars, 10 μm.
(H) HeLa cells were cultured in basal or FBS-depleted media for 10 h and processed by proximity ligation assay (PLA) using anti-FLAG and anti-LC3 Abs. Representative confocal micrographs and quantification of FLAG-CCR5 and endogenous LC3 interaction by PLA are shown. Plotted data represent mean ± SEM (n = 3 independent experiments, n = 5–7 randomly selected fields/condition, each containing ∼10 cells). One-tailed, paired Student’s t test ∗p < 0.05. Nuclei were counterstained with DAPI. Scale bars, 20 μM.
(I) ATG16WT and KO HeLa cells were cultured in basal or FBS-depleted media for 10 h in presence or absence of CCL5 (50 nM). Western blotting of FLAG-CCR5 and ATG16 and densitometry analyses of FLAG-CCR5 protein levels are shown. GAPDH was loading control. Plots represent mean ± SEM (n = 3 independent experiments). One-tailed, paired Student’s t test ∗p < 0.05, ∗∗p < 0.01, ns, not significant. See also Figures S7 and S8.
Discussion
本研究において、我々はCCL3、CCL4、CCL5、およびそれらの同族受容体であるCCR5を、ミクログリアと神経細胞間の新たな有害なクロストークのメディエーターとして同定し、mTORC1の活性化を介して神経細胞のオートファジーを抑制し、凝集しやすいタンパク質のクリアランスを障害することを明らかにした。興味深いことに、この軸の活性化は、HD発症前およびタウ病態マウスモデルの脳で観察され、この神経炎症メカニズムが疾患の初期段階で神経変性過程を促進することが示唆された。このことは、神経変性疾患における病理学的事象を、臨床症状が現れる前に理解することの重要性と課題を浮き彫りにしている。実際、今回われわれは、HDモデルにおいてmTORC1シグナル伝達が疾患経過の初期に亢進することを示した。一方、われわれは以前、同じHDモデルマウスの後期顕在期に出現した、大きなハンチンチン凝集体を有する神経細胞では、この経路が減弱することを報告した。これは、我々が細胞培養、マウスモデル、ヒトのサンプルで示したように、mTORC1がこれらの凝集体に隔離されたためである30。このように、後期の疾患細胞で見られる病理学的変化は、疾患の発生につながる重要な出来事を反映していない可能性がある。
ケモカインによるCCR5の活性化は、受容体の輸送には影響を与えないが、リサイクリングエンドソームでのオートファゴソーム形成を阻害する。これにより、オートファジーによるCCR5の分解が阻害され、CCR5が蓄積する(図S8)。言い換えれば、CCR5とケモカインとの結合は、mHTTまたはタウを発現している細胞において自己制御ループを活性化し、CCR5タンパク質レベルのアップレギュレーションをもたらす。注目すべきことに、Tet-Offタウオパチーモデルにおける我々の実験は、この自己増幅ループを断ち切るには変異タンパク質の除去で十分であることを示唆している。このメカニズムは、われわれの神経変性マウスモデルで観察されたCCR5神経細胞発現の増加を説明することができる。
以上の結果から、神経細胞のオートファジーを非細胞自律的にダウンレギュレートする活性化ミクログリアの役割に関する知見が得られた。この有害なミクログリアとニューロンのクロストークは、神経変性の初期に活性化され、自己持続的である可能性があるが、治療的介入ができる可能性がある。
参考文献
神経変性疾患におけるオートファジーの役割:細胞の恒常性維持と疾患発症に関わるメカニズム
(紹介者:医学群医学類MY)
医学群医療科学類の学生が、プログレスセミナーを行いました。他大学からの研究室見学のHOさんも参加してくださいました。遠方からご参加ありがとうございました!
