SCIENCE ADVANCES. 29 Nov 2023 Vol 9, Issue 48
GTF2I dosage regulates neuronal differentiation and social behavior in 7q11.23 neurodevelopmental disorders (GTF2I量は7q11.23神経発達障害における神経分化と社会的行動を制御する)
Alejandro López-Tobón 1 2 3, Reinald Shyti 1 2, Carlo Emanuele Villa 1 2, Cristina Cheroni 2 3, Patricio Fuentes-Bravo 1, Sebastiano Trattaro 1 2 3, Nicolò Caporale 2 3, Flavia Troglio 1 2 3, Erika Tenderini 1, Marija Mihailovich 1 2, Adrianos Skaros 1 3, William T Gibson 4, Alessandro Cuomo 1, Tiziana Bonaldi 1, Ciro Mercurio 5, Mario Varasi 5, Lucy Osborne 6, Giuseppe Testa 1 2 3
- Department of Experimental Oncology, European Institute of Oncology IRCCS, Via Adamello 16, 20139 Milan, Italy.
- 2Human Technopole, Viale Rita Levi-Montalcini 1, 20157 Milan, Italy.
- 3Department of Oncology and Hemato-Oncology, University of Milan, Milan, Italy.
- 4Department of Medical Genetics, University of British Columbia, Vancouver, BC, Canada.
- 5Experimental Therapeutics Program, FIRC Institute of Molecular Oncology Foundation (IFOM), 20139 Milan, Italy.
- 6Department of Medicine, University of Toronto, Toronto, ON, Canada.
Abstract
Copy number variations at 7q11.23 cause neurodevelopmental disorders with shared and opposite manifestations. Deletion causes Williams-Beuren syndrome featuring hypersociability, while duplication causes 7q11.23 microduplication syndrome (7Dup), frequently exhibiting autism spectrum disorder (ASD). Converging evidence indicates GTF2I as key mediator of the cognitive-behavioral phenotypes, yet its role in cortical development and behavioral hallmarks remains largely unknown. We integrated proteomic and transcriptomic profiling of patient-derived cortical organoids, including longitudinally at single-cell resolution, to dissect 7q11.23 dosage-dependent and GTF2I-specific disease mechanisms. We observed dosage-dependent impaired dynamics of neural progenitor proliferation, transcriptional imbalances, and highly specific alterations in neuronal output, leading to precocious excitatory neuron production in 7Dup, which was rescued by restoring physiological GTF2I levels. Transgenic mice with Gtf2i duplication recapitulated progenitor proliferation and neuronal differentiation defects alongside ASD-like behaviors. Consistently, inhibition of lysine demethylase 1 (LSD1), a GTF2I effector, was sufficient to rescue ASD-like phenotypes in transgenic mice, establishing GTF2I-LSD1 axis as a molecular pathway amenable to therapeutic intervention in ASD.
7q11.23のコピー数変異は、相反する症状を持つ神経発達障害を引き起こす。一方、重複は7q11.23マイクロ重複症候群(7Dup)を引き起こし、自閉スペクトラム症(ASD)をしばしば示す。GTF2Iが認知行動表現型の重要なメディエーターであることを示す証拠が集まっているが、大脳皮質の発達と行動の特徴におけるその役割はまだほとんどわかっていない。われわれは、患者由来の大脳皮質オルガノイドのプロテオミクスとトランスクリプトミクスのプロファイリングを統合し、単一細胞レベルでの縦断的解析も行い、7q11.23の用量依存的かつGTF2I特異的な疾患メカニズムを明らかにした。その結果、投与量に依存した神経前駆細胞増殖の動態障害、転写の不均衡、神経細胞出力の極めて特異的な変化が観察され、7Dupにおける興奮性神経細胞の早期産生につながったが、これは生理的なGTF2Iレベルを回復させることで回復した。Gtf2iが重複したトランスジェニックマウスでは、ASD様行動と同時に、前駆細胞の増殖と神経分化の欠陥が再現された。GTF2Iのエフェクターであるリジン脱メチラーゼ1(LSD1)の阻害は、トランスジェニックマウスにおけるASD様表現型の回復に十分であり、GTF2I-LSD1軸がASDの治療的介入に適した分子経路であることを立証した。
Introduction
7q11.23のコピー数変異は、相反する症状を持つ神経発達障害を引き起こす。一方、重複は7q11.23マイクロ重複症候群(7Dup)を引き起こし、自閉スペクトラム症(ASD)をしばしば示す。GTF2Iが認知行動表現型の重要なメディエーターであることを示す証拠が集まっているが、大脳皮質の発達と行動の特徴におけるその役割はまだほとんどわかっていない。われわれは、患者由来の大脳皮質オルガノイドのプロテオミクスとトランスクリプトミクスのプロファイリングを統合し、単一細胞レベルでの縦断的解析も行い、7q11.23の用量依存的かつGTF2I特異的な疾患メカニズムを明らかにした。その結果、投与量に依存した神経前駆細胞増殖の動態障害、転写の不均衡、神経細胞出力の極めて特異的な変化が観察され、7Dupにおける興奮性神経細胞の早期産生につながったが、これは生理的なGTF2Iレベルを回復させることで回復した。Gtf2iが重複したトランスジェニックマウスでは、ASD様行動と同時に、前駆細胞の増殖と神経分化の欠陥が再現された。GTF2Iのエフェクターであるリジン脱メチラーゼ1(LSD1)の阻害は、トランスジェニックマウスにおけるASD様表現型の回復に十分であり、GTF2I-LSD1軸がASDの治療的介入に適した分子経路であることを立証した。
26から28の遺伝子にまたがる7q11.23遺伝子座のコピー数変異(CNV)は、2つのまれな神経発達障害を引き起こし、多系統の表現型が共有され、正反対であることを特徴としている。ヘテロ接合体の欠失は、知的障害に加え、過社交性、不安、比較的よく保たれた言語能力を特徴とするウィリアムズ・ビューレン症候群[WBS; OMIM (Online Mendelian Inheritance in Man) 194050]を引き起こす(1)。7q11.23ヘテロ接合性重複(7Dup;OMIM 609757)は、知的障害と不安感を共有し、対照的に表現言語が障害され、自閉症スペクトラム障害(ASD)の発症率が高い(2, 3)。共有する臨床表現型と正反対の臨床表現型が多面的に組み合わさっていることから、両疾患の根底にある分子メカニズムも同様に、二律背反的あるいは類似している可能性が示唆される。それゆえ、神経精神症状と遺伝子病変のユニークな組み合わせを通して、7q11.23 CNV症候群は、社会的行動や言語能力に関与する遺伝子の用量脆弱性回路を解明する前例のない機会を提供している(4)。遺伝子の投与量と表現型の関連は、ゲノムの投与量感受性領域と特定の疾患とを結びつけ、CNVの特性を明らかにし、何千もの高投与量感受性遺伝子を同定した、ヒトの大規模サンプルを用いた最近の研究で強調されている(5)。
非典型的欠失を持つWBS患者を含むマウスとヒトの研究から得られた一致した証拠は、一般転写因子I(GTF2I)が7q11.23 CNVの認知行動学的発現の重要なドライバーであることを示している(6, 7)。GTF2Iはシグナル誘導転写因子であり、その標的遺伝子は、軸索誘導、カルシウムシグナル伝達、細胞周期、γ-アミノ酪酸(GABA)介在ニューロン(IN)の成熟など、さまざまな神経機能に関与している(8-10)。以前、私たちは、多能性状態におけるGTF2Iの用量依存的な転写制御異常を定義し、神経前駆細胞への分化に伴って増幅することを追跡した。その結果、GTF2Iはヒストンリジン脱メチル化酵素1(LSD1またはKDM1A)と結合することで転写を抑制すること、一方、LSD1を化学的および遺伝的に阻害することで、7Dup系においてGTF2Iの投与量増加によって生じた異常な転写抑制が解除され、知的障害やASDに関与する主要遺伝子の発現に影響を与え、LSD1阻害による治療介入の可能性が示された(11)。
患者由来の多能性幹細胞モデル、特に脳オルガノイドと単一細胞トランスクリプトミクスの併用は、これまでにない精度で精神神経疾患をモデル化する強力なプラットフォームである(12-14)。大脳皮質の発達や行動を制御する遺伝的回路など、ヒトでは他に類を見ないほど複雑なプロセスを研究するために、この手法を応用することは、発達過程で障害を受けやすい遺伝的制御のハブを発見する大きな可能性を秘めている。このようなアプローチの可能性を証明するものとして、ASDリスク遺伝子の発症への影響が、神経細胞分化のダイナミクスと神経発達の軌道の変化に収束することが、最近われわれや他の研究者によって示された(13, 15)。
ここでは、大脳皮質発達における7q11.23 CNVの影響について系統的な解析を行い、複数の個体、主要な発達時期、実験モデルにわたって、WBSと7Dupの転写および表現型ランドスケープを並置した。大脳皮質オルガノイド(CO)と13の人工多能性幹細胞(iPSC)株におけるシングルセル・トランスクリプトミクスを統合した結果、7q11.23遺伝子のドーサージ不均衡の結果として、前駆細胞の増殖と神経細胞の成熟のダイナミクスの変化が明らかになった。COまたはトランスジェニックマウスにおいて、GTF2Iの投与量を変化させるだけで、COで観察された主要な神経細胞分化の変化を表現するのに十分であり、GTF2IエフェクターであるLSD1を阻害することでASD様行動を引き起こすことがわかった。
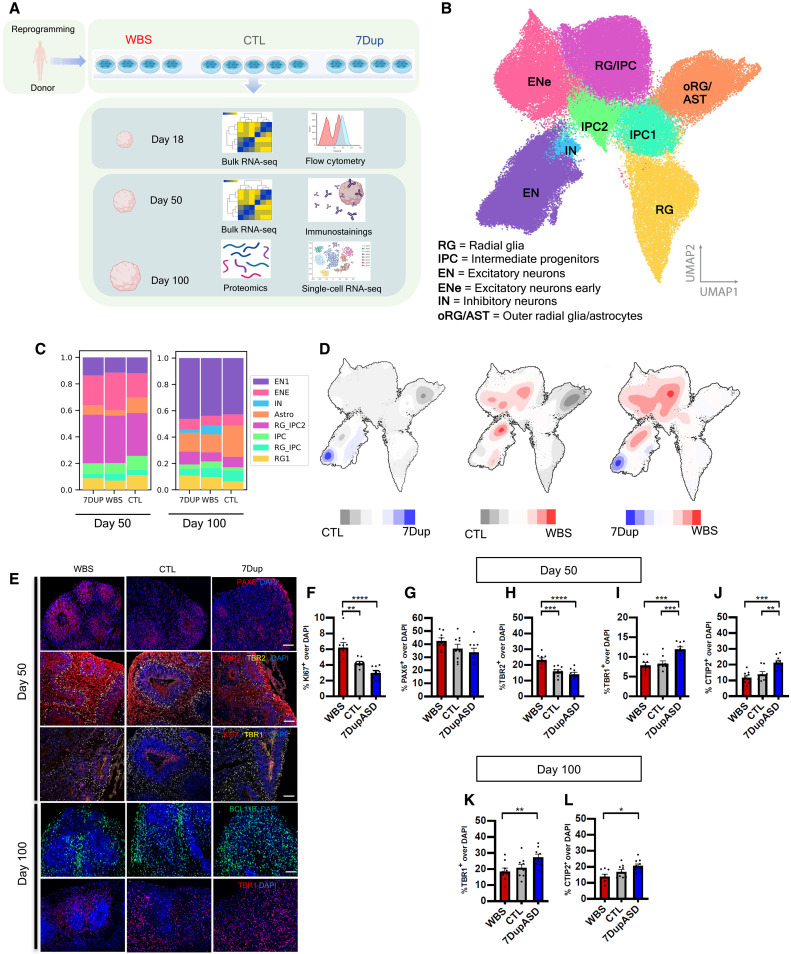
1A: 実験デザインの概略を示す図で、13のiPSCラインからCOを作成し、その発達をトランスクリプトミクスやシングルセルRNAシークエンスを用いて分析する過程が描かれている。
1B: 97,108個の単一細胞トランスクリプトームから導かれたUMAPを用いて、8つの異なる細胞クラスタを識別している。
1C: 分化50日目と100日目の各細胞クラスタ内の各遺伝子型の細胞割合を示すグラフである。
1D: UMAP上の細胞豊富度の違いを示す密度プロットで、WBS、7Dup、対照の各条件下での細胞豊富度の違いが色分けされている。
1E-J: 分化50日目のCOにおける異なる皮質集団のマーカーで免疫染色された画像と、それに対応する細胞割合の定量化を示している。
これらは、7q11.23のCNVが神経前駆細胞の増殖と分化のプロセス、及び神経細胞集団の出力に影響を与えることを示しており、特に7Dupでは早期段階で興奮性ニューロンの生産が促進されることが確認されている。
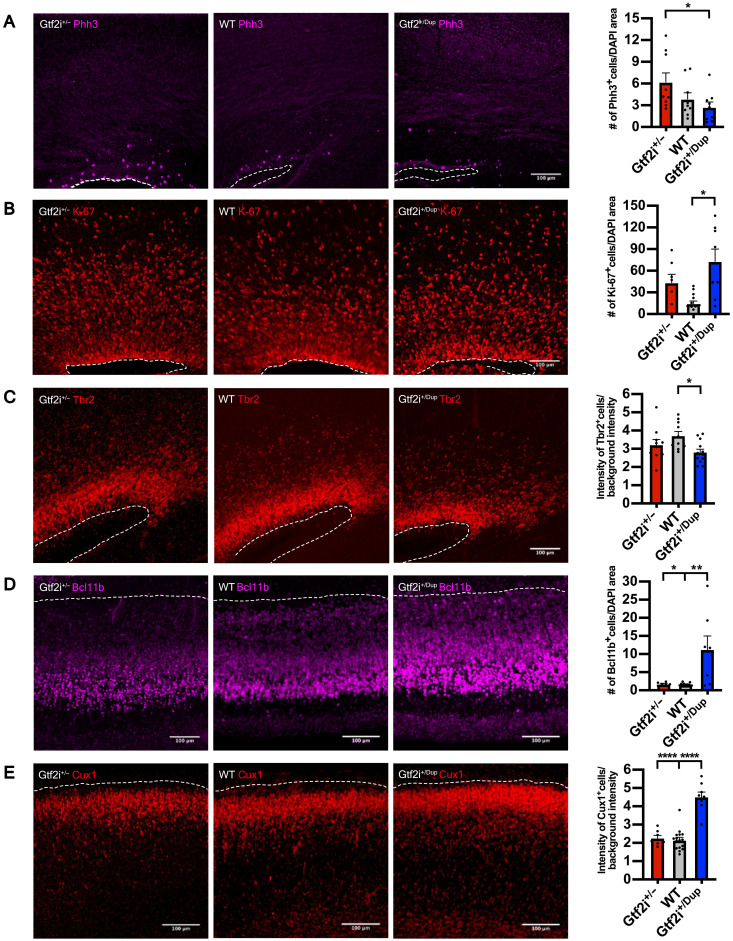
図5AとBは、胎児期のマウス脳における有糸分裂マーカーPhh3と細胞周期マーカーKi-67の発現を、Gtf2i遺伝子型別に示している。これは神経前駆細胞の増殖と神経分化における遺伝子用量の影響を示唆するものである。
図5Cは、中間前駆細胞マーカーTbr2の発現を示し、遺伝子用量による神経分化の変化が見られる。
図5DとEは、上層および下層皮質ニューロンマーカーBcl11bとCux1の発現を示し、神経分化の過程における遺伝子用量の影響を示している。
これらは、Gtf2i遺伝子の用量が神経前駆細胞の増殖と神経分化に影響を与える様子を示し、7q11.23コピー数変異に関連する神経発達障害の理解に寄与するものである。
Discussion
WBS症候群と7Dup症候群は、神経発達障害の中でも、CNVが正反対の表現型と正反対の表現型を持つ疾患の典型であり、社交性や言語といった人間の基礎的な側面の根底にある用量感受性メカニズムについて、ユニークな知見を与えてくれる(4)。しかし、このユニークな説明の可能性は、3つの主な限界のために、ほとんど満たされないままである。第一に、遺伝学的証拠によって、遺伝子の投与量とヒトの高次の表現型との間に明確な因果関係が示されている一方で、それらの関係を媒介する神経発達の先行要因は、まだほとんど解明されていない。それゆえ、発達の制約を強調し、典型的な発達と非典型的な発達の両方を、その根底にある軌跡の観点から解決する必要性を強調した神経構成主義の枠組みは、まさにWBSにその多くを負っているのだが、分子学的な用語ではまだ翻訳されていない(54)。第二に、このような因果関係を媒介する神経発達の先行因子を追跡するには、高次機能とその根底にある遺伝子の投与量との間の距離を埋めるために、複数の実験系を組み合わせる必要がある。しかし、これまでの研究では、動物モデルまたは2次元(2D)in vitro細胞モデルを個別に用いていたため、神経発達の軌跡を高解像度で追跡することができず、また、生物学的機能の異なる層で展開するエンドフェノタイプ間の橋渡しに必要な実験系間の交差検証もできなかった。第三に、7q11.23 CNVのシスエピスタシスは、CNV内の複数の遺伝子の同時投与量の不均衡が表現型に寄与する可能性があり、そのモデリングは悪名高く困難である。そのため、最近、疾患の病態生理に関連する表現型における7q11.23遺伝子座遺伝子の役割を明らかにすることに私たちや他の研究者が成功したにもかかわらず(11, 55-58)、7q11.23遺伝子の投与量変化が神経発達や行動発現に及ぼす具体的な影響は依然として不明である。ここでは、COとトランスジェニックマウスから得られた証拠を組み合わせて、GTF2Iとその軸が、複雑な行動発現の根底にある神経細胞成熟の重要なハブであることを明らかにした。
動物実験や個体群研究では、社交性の表現型においてGTF2Iが重要な役割を果たすとされてきた(4, 69-72)。一方、プロトカドヘリンはシナプス細胞接着分子であり、シナプスの構成と機能に重要な役割を果たしている(73, 74)。予想されるように、プロトカドヘリンに変異が生じると、神経細胞のコミュニケーションや機能に異常が生じ、ひいては社交性などの高次の認知過程に影響を及ぼす(75)。バルクRNAシーケンス(RNA-seq)の結果、7Dup COsおよびGtf2i+/Dupマウスにおいて、LSD1阻害後にプロトカドヘリンに変調をきたすことが明らかになり、社交性を媒介する用量感受性GTF2I -プロトカドヘリン分子軸について、種や実験モデルを超えて収束していることが示唆された。これらのデータから、7q11.23遺伝子の投与量変化が神経発達と行動表現型に及ぼす影響の包括的な分子学的特徴が明らかになり、GTF2I-LSD1軸が7q11.23疾患の病態生理の中心的なメディエーターであり、治療の可能性があることが明らかになった。
