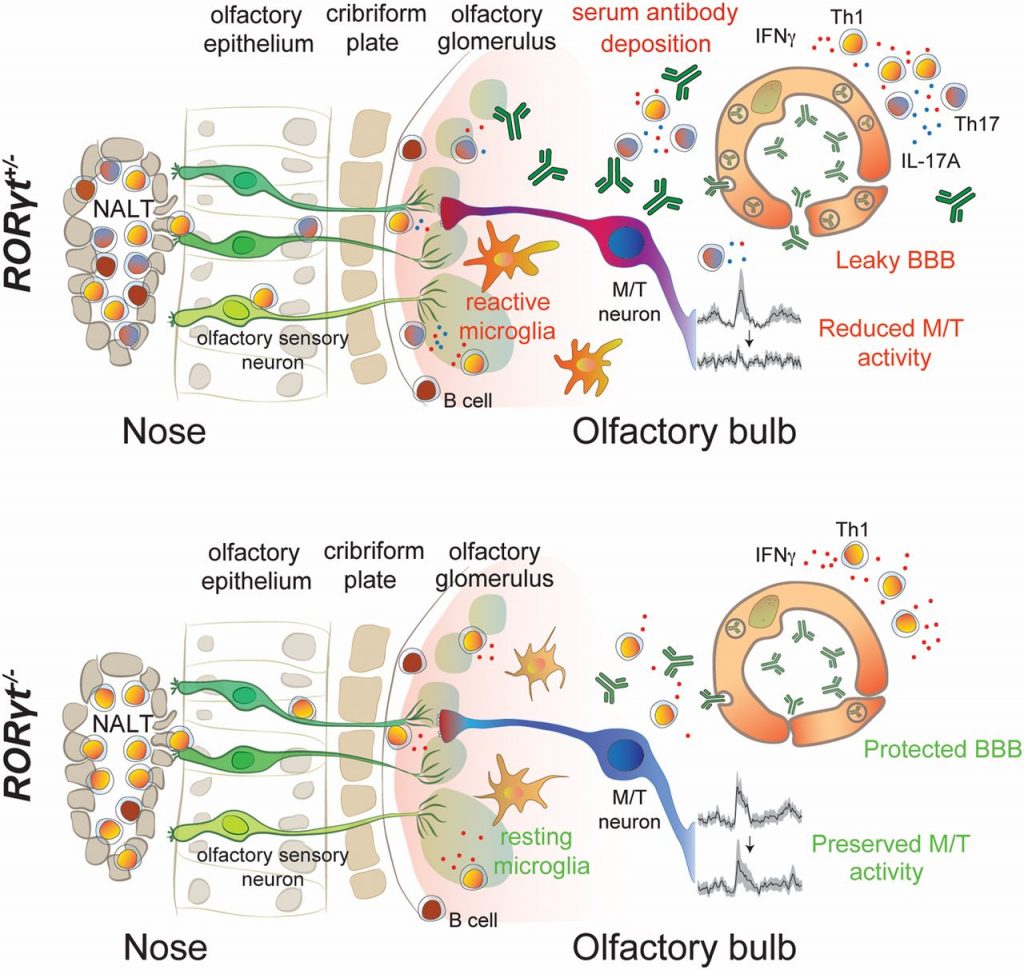
本日はJournal ClubでPNASに掲載された以下の論文が紹介されました。本論文では、グループA連鎖球菌による複数の感染によって引き起こされる感染後の自己免疫性脳炎のマウスモデルにおいて、Th17リンパ球がCNSへの自己抗体の侵入、持続的なミクログリアの活性化、および匂い処理における神経生理学的障害に重要であることを示しています。
RESEARCH ARTICLE
Th17 lymphocytes drive vascular and neuronal deficits in a mouse model of postinfectious autoimmune encephalitis (Th17リンパ球は感染後の自己免疫性脳炎のマウスモデルにおいて血管および神経細胞の欠損を引き起こす)
Maryann P. Platt, Kevin A. Bolding, Charlotte R. Wayne, Sarah Chaudhry, Tyler Cutforth, Kevin M. Franks, and Dritan AgalliuPNAS March 24, 2020 117 (12) 6708-6716; first published March 11, 2020 https://doi.org/10.1073/pnas.1911097117
- Edited by Lawrence Steinman, Stanford University School of Medicine, Stanford, CA, and approved February 14, 2020 (received for review June 27, 2019)
https://www.pnas.org/content/117/12/6708
Significance
Antibodies against neuronal receptors and synaptic proteins are associated with a group of ill-defined central nervous system (CNS) autoimmune diseases named autoimmune encephalitides (AE), characterized by an abrupt onset of seizures and movement and psychiatric deficits. How these antibodies enter the brain to trigger neuroinflammation, and how they affect the function of neural circuits, remains poorly understood. Here, we demonstrate that Th17 lymphocytes are critical for entry of autoantibodies into the CNS, persistent microglia activation, and neurophysiological deficits in odor processing, in a mouse model of postinfectious autoimmune encephalitis triggered by multiple infections with group A Streptococcus. Our findings emphasize the critical role that Th17 lymphocytes play in disease pathogenesis to impair CNS function in AE syndromes.
Abstract
Antibodies against neuronal receptors and synaptic proteins are associated with a group of ill-defined central nervous system (CNS) autoimmune diseases termed autoimmune encephalitides (AE), which are characterized by abrupt onset of seizures and/or movement and psychiatric symptoms. Basal ganglia encephalitis (BGE), representing a subset of AE syndromes, is triggered in children by repeated group A Streptococcus (GAS) infections that lead to neuropsychiatric symptoms. We have previously shown that multiple GAS infections of mice induce migration of Th17 lymphocytes from the nose into the brain, causing blood–brain barrier (BBB) breakdown, extravasation of autoantibodies into the CNS, and loss of excitatory synapses within the olfactory bulb (OB). Whether these pathologies induce functional olfactory deficits, and the mechanistic role of Th17 lymphocytes, is unknown. Here, we demonstrate that, whereas loss of excitatory synapses in the OB is transient after multiple GAS infections, functional deficits in odor processing persist. Moreover, mice lacking Th17 lymphocytes have reduced BBB leakage, microglial activation, and antibody infiltration into the CNS, and have their olfactory function partially restored. Th17 lymphocytes are therefore critical for selective CNS entry of autoantibodies, microglial activation, and neural circuit impairment during postinfectious BGE.
- autoimmune encephalitis
- blood–brain barrier
- Th17 lymphocyte
- postinfectious basal ganglia encephalitis
- olfactory circuitry